背景介绍
先天性肾上腺皮质增生症(CAH)是一种最常见的常染色体隐性遗传病,其经典(严重)形式可能危及生命,而非经典(轻微)形式可能无症状或导致女性不孕。CAH最常见的类型是21-羟化酶缺乏症;CAH是一种多种激素失衡的疾病。CYP21A2基因(编码21-羟化酶,一种细胞色素P-450酶)的突变导致缺乏21-羟化酶,而这种酶是肾上腺皮质中皮质醇和醛固酮生产所必需的。这种酶的缺乏会连锁反应。皮质醇的减少导致垂体促肾上腺皮质激素过度分泌,刺激皮质醇前体的积累,以及随后通过类固醇途径的转移,这些途径产生肾上腺雄激素。如今,经典形式是46,XX新生儿非典型生殖器官最常见的原因,也是儿童时期原发性肾上腺功能不足的主要原因。
CAH的疾病表型范围通常与CYP21A2基因型和每种基因型预期的残余21-羟化酶活性相关。基于激素的、拯救生命的新生儿筛查,针对经典形式的筛查始于1977年的阿拉斯加,现在已在美国所有50个州和全球40多个国家实施。 根据全球数百万新生儿的筛查数据,经典CAH的发生率在1/10,000到1/20,000的活产婴儿中。非经典CAH最早在1957年由法国生物化学家Jacques Decourt及其同事发现,而在20年后,对经典CAH患者的亲属的研究揭示了一些具有生化和遗传非经典CAH的人是无症状的,不需要治疗。非经典CAH在全球范围内很常见,估计的患病率在200人中有1例到1000人中有1例。与治疗其他形式的肾上腺功能不足的方法不同,CAH的治疗目标是双重的:首先,替代缺乏的激素;其次,减少过多的雄激素水平。尽管由于遗传学、代谢组学和治疗策略的进步,绝大多数CAH患者能够存活下来,但现有的治疗未能预防多种并存状况,肾上腺危象导致的死亡仍然发生。
$\sqrt{a}$
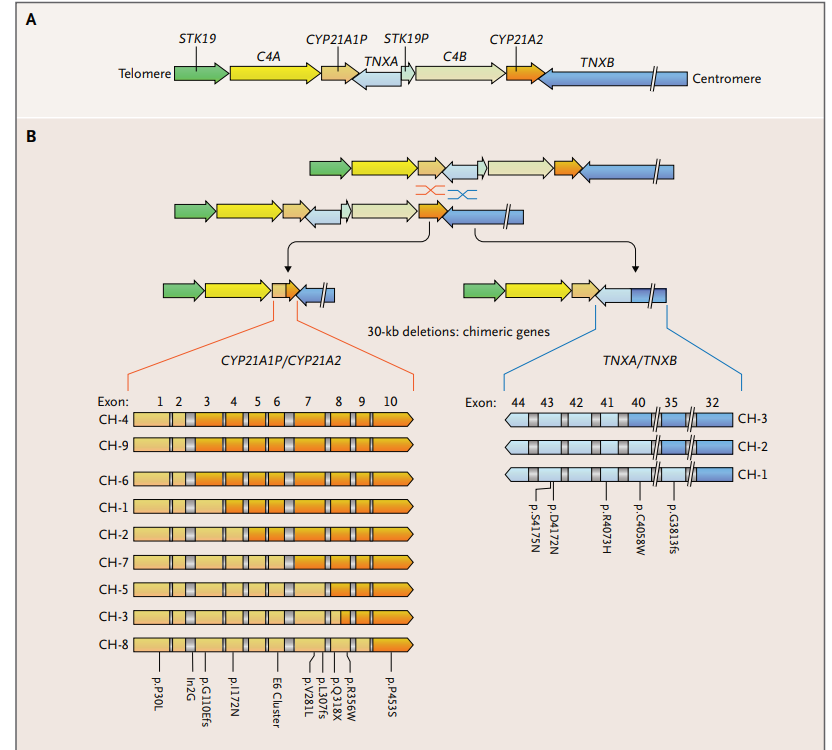
CYP21A2定位于第6号染色体(6p21.3),位于主要组织相容性复合体中的一个低拷贝重复序列位点,包括真基因和假基因(图1A)。已知有200多种CYP21A2突变;然而,大多数突变涉及10个有害变异,这些变异来源于非功能性的CYP21A1P,并通过同源重组在减数分裂期间的错位和基因转换产生。大约20%到30%的CYP21A2经典突变是30-kb的缺失,通常与空缺突变相关联。 然而,连接位点在临床上可能是相关的,因为大约3%的缺失由于连接位点的位置而保留了部分21-羟化酶活性,并与较温和的表型相关联17(图1B)。大多数患者是复合杂合,每个等位基因上有不同的突变,表型对应于较温和的基因缺陷。大约10%的CAH患者有CAH-X综合征,其特征是CAH的特征与高流动性型埃勒斯-当洛斯综合征的特征相结合,这是由于连续基因缺失破坏了CYP21A2和TNXB,通过基因分型诊断。TNXB编码的tenascin X是一种大型胞外基质蛋白,参与胶原沉积。大多数CAH-X综合征等位基因是由于单等位基因存在一个非功能性的TNXA/TNXB嵌合基因;双等位基因遗传导致更严重的症状。CAH-X的临床表型包括关节过度活动、关节痛、关节脱位、疝气和中线缺陷,其中包括心脏结构异常。
类固醇激素测量是诊断CAH的标准。由于CYP21A2位点的复杂性,二代测序不作为一线诊断测试。基因重复和缺失、CYP21A1P假基因以及一些等位基因中的多个突变使得很难将患者与携带者区分开来,通常需要父母的基因分型来确认基因型。
基因信息
CYP21A2 检测过程中的常见干扰就是来自其假基因 CYP21A1P 。1
2>NM_000500.9 CYP21A2 [organism=Homo sapiens] [GeneID=1589] [transcript=1]
>NR_040090.1 CYP21A1P[organism=Homo sapiens] [GeneID=1590]
基因信息可以直接NCBI上查询下载,在这里为了更方便的查看两个基因序列的相似度,在这贴一下两个基因多重比对的结果,如下:1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122NM_000500.9_CYP21A2_[organism=Ho ---------------------------------------------GTCTcGCCATGCTGC
NR_040090.1_CYP21A1P[organism=Ho ctggggctcttgagctataagtggcacctcagggccctgacgggcGTCTtGCCATGCTGC
NM_000500.9_CYP21A2_[organism=Ho TCCTGGGCCTGCTGCTGCTGCTGCCCCTGCTGGCTGGCGCCCGCCTGCTGTGGAACTGGT
NR_040090.1_CYP21A1P[organism=Ho TCCTGGGCCTGCTGCTGCTGCTGCCCCTGCTGGCTGGCGCCCGCCTGCTGTGGAACTGGT
NM_000500.9_CYP21A2_[organism=Ho GGAAGCTCCGGAGCCTCCACCTCCcGCCTCTTGCCCCGGGCTTCTTGCACCTGCTGCAGC
NR_040090.1_CYP21A1P[organism=Ho GGAAGCTCCGGAGCCTCCACCTCCtGCCTCTTGCCCCGGGCTTCTTGCACCTGCTGCAGC
NM_000500.9_CYP21A2_[organism=Ho CCGACCTCCCCATCTATCTGCTTGGCCTGACTCAGAAATTCGGGCCCATCTACAGGCTCC
NR_040090.1_CYP21A1P[organism=Ho CCGACCTCCCCATCTATCTGCTTGGCCTGACTCAGAAATTCGGGCCCATCTACAGGCTCC
NM_000500.9_CYP21A2_[organism=Ho ACCTTGGGCTGCA-----------------------------------------------
NR_040090.1_CYP21A1P[organism=Ho ACCTTGGGCTGCAaggtgagaggctgatctcgctctggccctcaccataggagggggcgg
NM_000500.9_CYP21A2_[organism=Ho --------------------------------------------------AGATGTGGTG
NR_040090.1_CYP21A1P[organism=Ho aggtgacggagagggtcctctctccgctgacgctgctttggctgtctcccAGATGTGGTG
NM_000500.9_CYP21A2_[organism=Ho GTGCTGAACTCCAAGAGGACCATTGAGGAAGCCATGGTCAAAAAGTGGGCAGACTTTGCT
NR_040090.1_CYP21A1P[organism=Ho GTGCTGAACTCCAAGAGGACCATTGAGGAAGCCATGGTCAAAAAGTGGGCAGACTTTGCT
NM_000500.9_CYP21A2_[organism=Ho GGCAGACCTGAGCCACTTACCT--------------------------------------
NR_040090.1_CYP21A1P[organism=Ho GGCAGACCTGAGCCACTTACCTgtaagggccgggggcattttttctttcttaaacaaatt
NM_000500.9_CYP21A2_[organism=Ho ------------------------------------------------------------
NR_040090.1_CYP21A1P[organism=Ho ttttttttgttagagatggggtcttgctatgttgcccaggctggtcttgaattcctggtc
NM_000500.9_CYP21A2_[organism=Ho ------------------------------------------------------------
NR_040090.1_CYP21A1P[organism=Ho tcaagtgatcctcccacctcggcctcaagtgggagccaccttcgggggcttccccaatcc
NM_000500.9_CYP21A2_[organism=Ho ------------------------------------------------------------
NR_040090.1_CYP21A1P[organism=Ho tccaggtcactggaagctcttggggggcatatcttcaggagaagaagcaggtgttgagga
NM_000500.9_CYP21A2_[organism=Ho ------------------------------------------------------------
NR_040090.1_CYP21A1P[organism=Ho ggcagaagaaggtcaggccctcggcttccttggtcagttcccaccctccagcccccagct
NM_000500.9_CYP21A2_[organism=Ho ----------ACAAGCTGGTGTCTAgGAACTACCCGGACCTGTCcTTGGgagactacTCc
NR_040090.1_CYP21A1P[organism=Ho cctcctgcagACAAGCTGGTGTCTAaGAACTACCCGGACCTGTCgTTGG--------TCt
NM_000500.9_CYP21A2_[organism=Ho CTGCTCTGGAAAGCCCACAAGAAGCTCACCCGCTCAGCCCTGCTGCTGGGCATCCGTGAC
NR_040090.1_CYP21A1P[organism=Ho CTGCTCTGGAAAGCCCACAAGAAGCTCACCCGCTCAGCCCTGCTGCTGGGCATCCGTGAC
NM_000500.9_CYP21A2_[organism=Ho TCCATGGAGCCAGTGGTGGAGCAGCTGACCCAGGAGTTCTGTGAGCGCATGAGAGCCCAG
NR_040090.1_CYP21A1P[organism=Ho TCCATGGAGCCAGTGGTGGAGCAGCTGACCCAGGAGTTCTGTGAGCGCATGAGAGCCCAG
NM_000500.9_CYP21A2_[organism=Ho CCCGGCACCCCTGTGGCCATTGAGGAGGAATTCTCTCTCCTCACCTGCAGCATCAtCTGT
NR_040090.1_CYP21A1P[organism=Ho CCCGGCACCCCTGTGGCCATTGAGGAGGAATTCTCTCTCCTCACCTGCAGCATCAaCTGT
NM_000500.9_CYP21A2_[organism=Ho TACCTCACCTTCGGAGACAAGATCAAGGAcGACAACTTAATGCCTGCCTATTACAAATGT
NR_040090.1_CYP21A1P[organism=Ho TACCTCACCTTCGGAGACAAGATCAAGGAgGACAACTTAATGCCTGCCTATTACAAATGT
NM_000500.9_CYP21A2_[organism=Ho ATCCAGGAGGTGTTAAAAACCTGGAGCCACTGGTCCATCCAAATTGTGGACGTGATTCCC
NR_040090.1_CYP21A1P[organism=Ho ATCCAGGAGGTGTTAAAAACCTGGAGCCACTGGTCCATCCAAATTGTGGACGTGATTCCC
NM_000500.9_CYP21A2_[organism=Ho TTTCTCAGGTTCTTCCCCAATCCAGGTCTCCGGAGGCTGAAGCAGGCCATAGAGAAGAGG
NR_040090.1_CYP21A1P[organism=Ho TTTCTCAGGTTCTTCCCCAATCCAGGTCTCCGGAGGCTGAAGCAGGCCATAGAGAAGAGG
NM_000500.9_CYP21A2_[organism=Ho GAtCACAtCGtGGAGAtGCAGCTGAGGCAGCACAAGGAGAGCCTcGTGGCAGGCCAGTGG
NR_040090.1_CYP21A1P[organism=Ho GAcCACAaCGaGGAGAaGCAGCTGAGGCAGCACAAGGAGAGCCTgGTGGCAGGCCAGTGG
NM_000500.9_CYP21A2_[organism=Ho AGGGACATGATGGACTACATGCTCCAAGGGGTGGCGCAGCCGAGCATGGAAGAGGGCTCT
NR_040090.1_CYP21A1P[organism=Ho AGGGACATGATGGACTACATGCTCCAAGGGGTGGCGCAGCCGAGCATGGAAGAGGGCTCT
NM_000500.9_CYP21A2_[organism=Ho GGACAGCTCCTGGAAGGGCACgTGCACATGGCTGCAGTGGACCTCCTGATCGGTGGCACT
NR_040090.1_CYP21A1P[organism=Ho GGACAGCTCCTGGAAGGGCACtTGCACATGGCTGCAGTGGACCTCCTGATCGGTGGCACT
NM_000500.9_CYP21A2_[organism=Ho GAGACCACAGCAAACACCCTCTCCTGGGCCGTGG-TTTTTTTGCTTCACCACCCTGAGAT
NR_040090.1_CYP21A1P[organism=Ho GAGACCACAGCAAACACCCTCTCCTGGGCCGTGGtTTTTTTTGCTTCACCACCCTGAGAT
NM_000500.9_CYP21A2_[organism=Ho TCAGCAGCGACTGcAGGAGGAGCTAGACCACGAACTGGGCCCTGGTGCCTCCAGCTCCCG
NR_040090.1_CYP21A1P[organism=Ho TCAGCAGCGACTGtAGGAGGAGCTAGACCACGAACTGGGCCCTGGTGCCTCCAGCTCCCG
NM_000500.9_CYP21A2_[organism=Ho GGTCCCCTACAAGGACCGTGCACGGCTGCCCTTGCTCAATGCCACCATCGCCGAGGTGCT
NR_040090.1_CYP21A1P[organism=Ho GGTCCCCTACAAGGACCGTGCACGGCTGCCCTTGCTCAATGCCACCATCGCCGAGGTGCT
NM_000500.9_CYP21A2_[organism=Ho GCGCCTGcGGCCCGTTGTGCCCTTAGCCTTGCCCCACCGCACCACACGGCCCAGCAGCAT
NR_040090.1_CYP21A1P[organism=Ho GCGCCTGtGGCCCGTTGTGCCCTTAGCCTTGCCCCACCGCACCACACGGCCCAGCAGCAT
NM_000500.9_CYP21A2_[organism=Ho CTCCGGCTACGACATCCCTGAGGGCACAGTCATCATTCCGAACCTCCAAGGCGCCCACCT
NR_040090.1_CYP21A1P[organism=Ho CTCCGGCTACGACATCCCTGAGGGCACAGTCATCATTCCGAACCTCCAAGGCGCCCACCT
NM_000500.9_CYP21A2_[organism=Ho GGATGAGACGGTCTGGGAGAGGCCACATGAGTTCTGGCCTGATCGCTTCCTGGAGCCAGG
NR_040090.1_CYP21A1P[organism=Ho GGATGAGACGGTCTGGGAGAGGCCACATGAGTTCTGGCCTGATCGCTTCCTGGAGCCAGG
NM_000500.9_CYP21A2_[organism=Ho CAAGAACTCCAGAGCTCTGGCCTTCGGCTGCGGTGCCCGCGTGTGCCTGGGCGAGCCGCT
NR_040090.1_CYP21A1P[organism=Ho CAAGAACTCCAGAGCTCTGGCCTTCGGCTGCGGTGCCCGCGTGTGCCTGGGCGAGCCGCT
NM_000500.9_CYP21A2_[organism=Ho GGCGCGCCTGGAGCTCTTCGTGGTGCTGACCCGACTGCTGCAGGCCTTCACGCTGCTGCC
NR_040090.1_CYP21A1P[organism=Ho GGCGCGCCTGGAGCTCTTCGTGGTGCTGACCCGACTGCTGCAGGCCTTCACGCTGCTGCC
NM_000500.9_CYP21A2_[organism=Ho CTCCGGGGACGCCCTGCCCTCCCTGCAGCCCCTGCCCCACTGCAGTGTCATCCTCAAGAT
NR_040090.1_CYP21A1P[organism=Ho CTCCGGGGACGCCCTGCCCTCCCTGCAGCCCCTGCCCCACTGCAGTGTCATCCTCAAGAT
NM_000500.9_CYP21A2_[organism=Ho GCAGCCTTTCCAAGTGCGGCTGCAGCCCCGGGGGATGGGGGCCCACAGCCCgGGCCAGAg
NR_040090.1_CYP21A1P[organism=Ho GCAGCCTTTCCAAGTGCGGCTGCAGCCCCGGGGGATGGGGGCCCACAGCCCaGGCCAGAa
NM_000500.9_CYP21A2_[organism=Ho CCAGTGATGGGGCAGGACCGATGCCAGCCGGGTACCTCAGTTTCTCCTTTATTGCTCCCG
NR_040090.1_CYP21A1P[organism=Ho CCAGTGATGGGGCAGGACCGATGCCAGCCGGGTACCTCAGTTTCTCCTTTATTGCTCCCG
NM_000500.9_CYP21A2_[organism=Ho TACGAACCCCTCCCCTCCCCCCTGTAAACACAGTGCTGCGAGATCGCTGGCAGAGAAGGC
NR_040090.1_CYP21A1P[organism=Ho TACGAACCCCTCCCCTCCCCCCTGTAAACACAGTGCTGCGAGATCGCTGGCAGAGAAGGC
NM_000500.9_CYP21A2_[organism=Ho TTCCTCCAGCGGCTGGGTGGTGAAGGACCCTGGCTCTTCTCTCGGGGCGACCCCTCAGTG
NR_040090.1_CYP21A1P[organism=Ho TTCCTCCAGCGGCTGGGTGGTGAAGGACCCTGGCTCTTCTCTCGGGGCGACCCCTCAGTG
NM_000500.9_CYP21A2_[organism=Ho CTCGGCAGTCATACTGGGGTGCGAGAGAGGTGGGCAGCAGCTCAGCCTCCCCCCGCTGGG
NR_040090.1_CYP21A1P[organism=Ho CTCGGCAGTCATACTGGGGTGCGAGAGAGGTGGGCAGCAGCTCAGCCTCCCCCCGCTGGG
NM_000500.9_CYP21A2_[organism=Ho GAGCGAAAGTTTCTTGGTCTCAGCTTCATTTCCGTGAAGGGCACCGAGAACTCGAAGCCC
NR_040090.1_CYP21A1P[organism=Ho GAGCGAAAGTTTCTTGGTCTCAGCTTCATTTCCGTGAAGGGCACCGAGAACTCGAAGCCC
NM_000500.9_CYP21A2_[organism=Ho TTCCAGTGGTACCAGCTCACTCCCTGGGAAAGGGGTTGTCAAGAGAGAGTCAAAGCCGGA
NR_040090.1_CYP21A1P[organism=Ho TTCCAGTGGTACCAGCTCACTCCCTGGGAAAGGGGTTGTCAAGAGAGAGTCAAAGCCGGA
NM_000500.9_CYP21A2_[organism=Ho TGTCCCATCTGCTCtTCCCGTTCCCCTTAAGGAGGTaGCTCCCAGCACTCAACCAACCTC
NR_040090.1_CYP21A1P[organism=Ho TGTCCCATCTGCTCcTCCCGTTCCCCTTAAGGAGGTgGCTCCCAGCACTCAACCAACCTC
NM_000500.9_CYP21A2_[organism=Ho CCCGCAGAGCTCCCTTCCTGACCCTCcGCtGCAGAGGATTGAGGCTTAATtCTGAGCTGG
NR_040090.1_CYP21A1P[organism=Ho CCCGCAGAGCTCCCTTCCTGACCCTCtGCcGCAGAGGATTGAGGCTTAATcCTGAGCTGG
NM_000500.9_CYP21A2_[organism=Ho cCCTTTCCAGCCAATAAATCAACTCCAGCTCCCTCTG
NR_040090.1_CYP21A1P[organism=Ho tCCTTTCCAGCCAATAAATCAACTCCAGCTCCCTCTG
references
[1]. Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency