在多重 PCR 引物设计中,香农熵(Shannon entropy) 用来量化多序列比对每一位上碱基(及空位)的多样性,从而客观找出保守区——适合设计通用引物或简并引物的区段。
与仅看「一致性序列」相比,香农熵对大量、异质性高的模板更敏感,也便于在软件里用统一数值阈值做自动化筛选。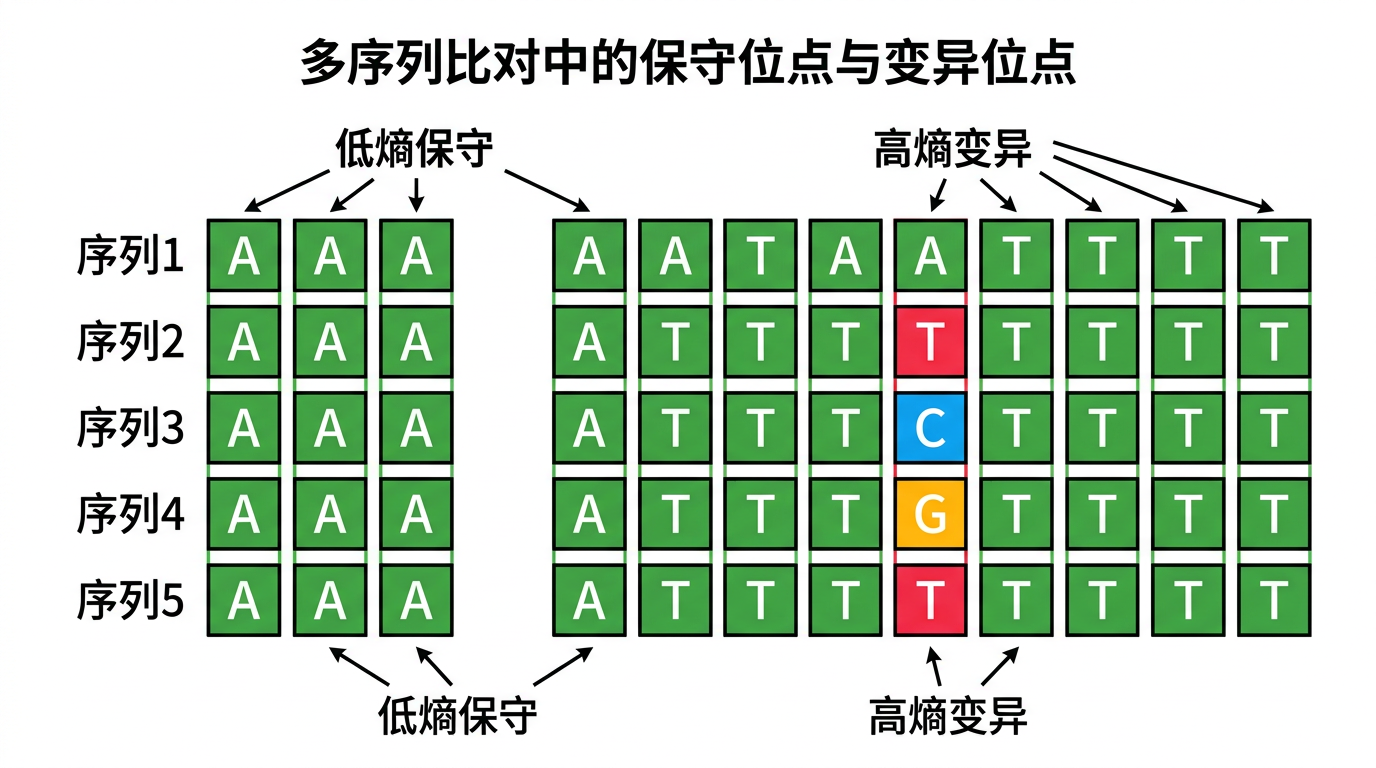
为什么需要香农熵
- 目标:在一条基因的多条同源序列上,找到跨序列尽量一致的片段,以便引物能稳定结合。
- 难点:序列条数多、变异分散时,肉眼扫「哪一段更保守」主观且易漏。
- 香农熵的作用:把每一位的「多样程度」压成一个数 (S),越小越保守,便于排序、滑动窗口、设阈值。
直观理解:低熵 vs 高熵
| 情况 | 比对位点上的符号 | 直觉 | 对引物设计 |
|---|---|---|---|
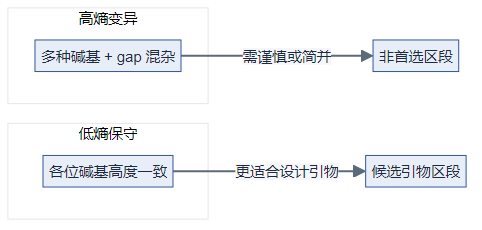
| 低熵 | 几乎全是同一种碱基(如全是 A) | 「很整齐」 | 优先选作引物结合区 |
| 高熵 | A/T/C/G 甚至 gap 混杂且较均匀 | 「很乱」 | 尽量避开或拆成简并方案 |
 |
1 | flowchart LR |
核心概念与原理
香农熵来自信息论,衡量不确定性。在比对语境下:
- 熵高:该位点可能出现的符号种类多、分布相对均匀 → 变异大,不确定性高。
- 熵低:某一种符号占绝对多数 → 保守,不确定性低。
因此,多重 PCR 希望在全长比对上找到 熵值持续较低 的连续窗口,作为引物落点。
在引物设计流程中的应用
将香农熵用于多重 PCR,通常与 MSA → 滑窗/合并保守区 → 设计引物 → 评估 的流程结合,可借助 PMPrimer、openPrimeR 等工具。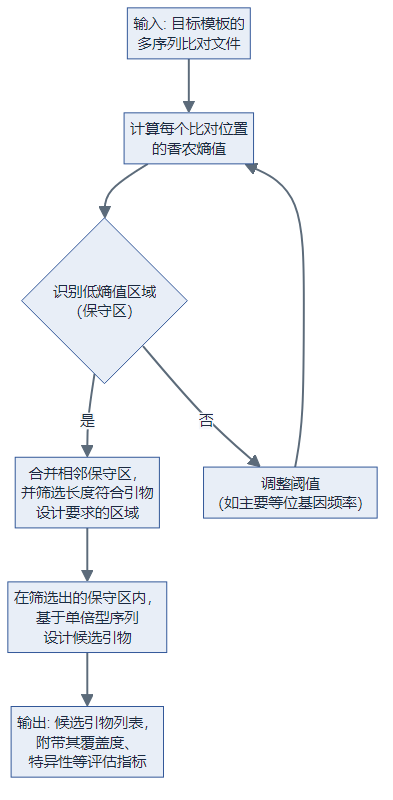
1 | flowchart TD |
如何计算与解读
在引物相关工具中,香农熵(有的界面记为 e)通常对 A、T、C、G 与 gap(-) 五类符号分别统计频率;将 gap 纳入可避免把「插入缺失」误判为保守。
计算公式
对某一比对位置,若共有 (n) 种符号((n \le 5):A/T/C/G/-),第 (i) 种频率为 (p_i),则香农熵(以 2 为底,单位 bit)为:
$$S = - \sum_i p_i \log_2(p_i)$$
当某 (p_i=0) 时,该项按惯例取 0(极限意义下 (p\log p \to 0))。
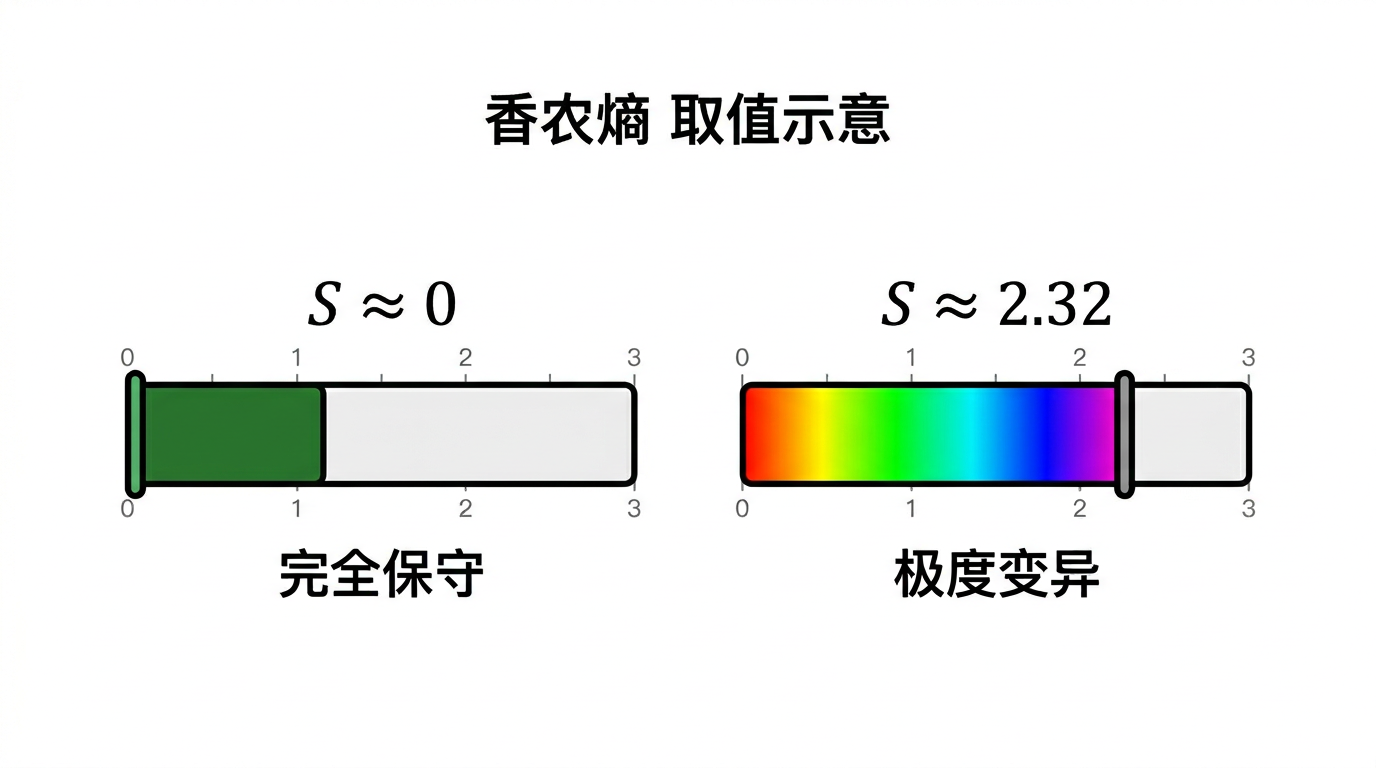
取值范围与直觉锚点
| (S) 大致范围 | 含义 |
|---|---|
| (S = 0) | 该位点完全保守,所有序列同一符号(最理想作引物结合位)。 |
| (0 < S < 2.32) | 存在一定变异;越大越乱,引物跨越时越需简并或避开。 |
| (S \approx 2.32) | A/T/C/G/- 五种各约 20% 时接近该上界(极度变异,极难用单一普通引物覆盖)。 |
说明:若某工具不把 gap 计入 (n),理论最大值会略低于 2.32,阅读软件文档时注意其定义。
文中的极简数值例(与原文一致)
| 序列 | Position 1 | Position 2 |
|---|---|---|
| Seq 1 | A | A |
| Seq 2 | A | T |
| Seq 3 | A | C |
| Seq 4 | A | G |
| Seq 5 | A | - (gap) |
- Position 1:全为 A,(p_A=1) → (S=0),完全保守。
- Position 2:A、T、C、G、- 各出现 1 次,(p=0.2) → (S = -5 \times 0.2 \times \log_2(0.2) \approx 2.32),极度变异。
阈值设定(与主要等位基因频率)
实践中常把「多保守算保守」转成可调参数:
- 例如设置主要等位基因最低频率(如 0.95):该位点上频率最高的碱基(或 gap)占比须 ≥95%,才视为足够保守。
- 工具据此换算成对应的香农熵阈值(如约 0.12),再对连续区段的平均熵或逐位熵做筛选(以软件说明为准)。
注意:阈值是实验与数据依赖的;属间/基因间保守性差异大时,宜在验证集上微调。
实践要点清单
- 比对质量:空位、比对算法会影响熵;同一批数据应用同一 MSA 流程。
- 窗口长度:单点低熵不够,通常要 连续窗口 平均熵低于阈值才作为引物区。
- 与简并引物衔接:高熵位点若无法避开,可结合 IUPAC 简并碱基 与覆盖度评估。
- 报告记录:写明 MSA 来源、序列条数、熵定义(是否含 gap)、阈值。
实例验证(文献与工具案例)
基于香农熵筛选保守区的方法已在复杂数据集上验证,例如 PMPrimer 相关报道中的:
- 葡萄球菌属 tuf 基因:2547 条序列、54 物种。
- 分枝杆菌科 hsp65:6528 条序列、多物种背景。
- 古菌 16S rRNA:11,757 条序列、跨多分类等级。
说明在万级序列、保守性参差的场景下,熵辅助仍有助于稳定定位可设计区。
总结
香农熵把「保守性」变成可比较、可阈值化的数值,便于与自动化流程结合:从海量比对中筛出低熵连续区,再进入引物设计与特异性评估,从而提升多重 PCR 在多样本模板上的成功率与可重复性。
延伸阅读
- 与 简并引物、IUPAC 符号 的配合见《引物设计-概述-引物分类》。