1. 文献信息
- 题目:DeepEnzyme: a robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D structures
- 期刊:Briefings in Bioinformatics,2024 年 8 月;卷 25 期 5,文章号 bbae409
- DOI:https://doi.org/10.1093/bib/bbae409
- 开放获取:PMC PMC11880767,PMID 39162313
- 预印本:bioRxiv 10.1101/2023.12.09.570923(2023-12)
- 通讯作者:上海交通大学 卢洪忠(hongzhonglu@sjtu.edu.cn)等
周转数(turnover number,kcat) 表示酶在底物饱和条件下、单位时间内每个活性位点所能转化的底物分子数上限,是蛋白工程、合成生物学与基因组尺度代谢模型(genome-scale metabolic model,GEM) 参数化的关键动力学量之一。
段末注释:kcat 为催化常数/周转数;GEM 指覆盖全基因组反应与酶学约束的代谢网络模型。
2. 研究动机与定位
实验测定 kcat 成本高、通量有限;深度学习可在序列或反应层面做高通量推断。已有工作包括:基于机器学习、主要面向大肠杆菌的 Heckmann 等;面向广谱酶–底物对的 DLKcat(Li 等,Nature Catalysis 2022);在底物/产物指纹与精炼数据上改进的 TurNuP(Kroll 等,Nat Commun 2023)等。作者指出:当测试集与训练集酶序列相似度较低时,纯序列模型的精度与稳健性仍不足。
蛋白质功能在很大程度上由三维结构决定;活性位点排布、底物结合与稳定性等信息难以仅从一级序列完全恢复。AlphaFold2、ColabFold 等降低了获得高质量结构的成本,使得在 kcat 预测中系统利用三维结构特征成为可能。DeepEnzyme 的定位是:监督学习框架下,显式融合 1D 序列、3D 结构与底物表征,以提升精度并在低序列相似度场景下更稳健。
3. 原文 Methods 与模型结构(详解)
以下按 Wang 等 Brief Bioinform 2024 正文 Methods 顺序整理,并在 §3.11 补充与 GitHub 仓库 示例代码可对齐的实现细节(论文未逐条列出超参数时,以代码与补充材料为准)。
3.1 数据预处理与划分(Data preprocessing)
- 数据来源:初始集合来自 DLKcat 论文发布的酶–底物–kcat 数据。
- 序列去冗余:用 MMseqs2 在全数据上评估酶序列相似度。对同一底物且两酶序列相似度 > 90% 的多条记录,只保留其中酶序列最长的那一条,以降低训练/验证/测试之间因近重复序列带来的过拟合风险。
- 规模:自原数据约 16 838 对酶–底物对,经上述规则得到 11 927 条独特组合(原文 Supplementary Table 2)。
- 划分:按 80% / 10% / 10% 随机划分为训练集、验证集与测试集。
段末注释:MMseqs2 为面向海量序列的快速同源搜索与聚类工具(Steinegger & Söding,Nat Biotechnol 2017),此处用于相似度筛选与去冗余。
3.2 酶结构获取与接触图(Enzyme structure prediction and contact map)
- 结构来源:数据集中大量酶无实验解析结构,作者使用 ColabFold 为全部酶预测三维结构;全文预测结构的平均 pLDDT 约为 92.67,作为结构质量的整体说明。
- 从坐标到图:将每条结构转为无向图形式的接触图。每个氨基酸残基对应图上的一个节点;若两残基「代表原子」之间的欧氏距离 小于给定阈值,则在两节点间连一条边,从而得到可供 GCN 使用的邻接关系。论文将具体构图准则指向文献 [44,45](结构导向的图神经网络在蛋白上的常见设定)。
- 与公开代码一致的一种实现:官方示例脚本
luciferase_contact_map中,对每个残基取 Cα(alpha carbon,Cα) 坐标,计算残基间 Cα–Cα 距离矩阵;以 10 Å(ångström,Å) 为截断,距离小于阈值则记为接触(二值化邻接矩阵),并对序列–残基数不一致等情况做填充与对角线处理。注意:该阈值为代码显式写出;若复现论文结果,应以作者提供的预处理流水线与训练配置为准。
段末注释:Cα 为主链上每个氨基酸的 α 碳原子,常用于残基几何中心近似;Å 为 10⁻¹⁰ m,为结构生物学常用长度单位。
3.3 序列分支:Transformer 与 n-gram(The protein sequence baseline)
- n-gram 词表化:与 DLKcat 中蛋白质序列处理思路一致,将氨基酸序列切分为长度为 n 的重叠子串(n-gram),经 词嵌入(word embedding) 将每个 n-gram 映为向量;不在词表中的片段在实现中可动态赋默认索引(见仓库
split_sequence)。 - Transformer 编码器:嵌入序列后接 位置编码(positional encoding) 与 Transformer 编码器;其中 多头自注意力(multi-head self-attention,MHSA) 通过 Query(Q)、Key(K)、Value(V)计算上下文相关表示(原文 Eq. 1–3,形式与 Vaswani 等经典定义同族)。
- 输出:得到与序列长度对齐的隐向量序列,作为序列模态特征,供后续与结构、底物特征融合。
段末注释:MHSA 为并行多组自注意力,再拼接/投影,以捕获不同子空间的依赖关系。
3.4 底物分支:SMILES、指纹与分子图 GCN(Substrate / RDKit)
- 分子解析:由底物 SMILES 用 RDKit 构建分子对象,并可加氢(
AddHs)以符合化学信息学惯例。 - 原子与键类型编码:原子类型、键类型等离散符号映射为整数索引(仓库中以
atom_dict、bond_dict等 pickle 词典提供)。 - 子结构指纹:在半径 radius 迭代下聚合邻居信息,形成层次化分子指纹向量(实现类 Morgan/环境展开思路,见
extract_fingerprints);指纹元素再嵌入为稠密向量。 - 邻接矩阵:由 RDKit 得到分子 邻接矩阵(adjacency matrix) 作为图结构,输入 GCN 分支;该分支与酶结构分支共用 GCN 模块类,但权重不共享,以分别学习底物图与蛋白接触图。
3.5 结构分支:接触图上的 GCN(Protein structure baseline)
- 将 §3.2 得到的蛋白邻接矩阵(稀疏或稠密形式)与序列分支中同一序列对应的残基级嵌入结合:对每个节点有初始特征向量,经 GCN 做邻域聚合(论文 Eq. 4;实现中常采用对称归一化邻接矩阵 D̃−1/2 Ã D̃−1/2 形式的图卷积,与 Kipf & Welling 经典 GCN 一致),输出更新后的节点/图表示,作为结构模态特征。
3.6 融合、神经注意力与 kcat 读出(Neural attention baseline)
- 多路特征汇合:原文将来自 序列 Transformer、蛋白接触图 GCN、底物图 GCN 的表示合并为一条综合嵌入(图示为「酶 + 底物」联合表征)。
- 神经注意力:在融合后使用神经注意力机制(原文 Eq. 5–8),对各子块隐向量赋权并聚合,经 整流线性单元(rectified linear unit,ReLU) 等非线性变换后,由线性层映射到标量输出。论文指出该设计可得到与残基相关的注意力权重,用于后续解释哪些位点对预测贡献更大。
- 标签空间(实现细节):官方推理示例将网络输出视为 log2(kcat)(或与之等价的对数刻度),再经
2**prediction还原为 kcat;训练阶段损失函数在正文以指标反推为主,具体损失形式以仓库训练脚本为准。
段末注释:ReLU 为 (\max(0,x)) 激活;公开仓库中前向网络可能与正文图示在注意力实现细节上略有差异,以论文公式与作者发布版本为准。
3.7 评价指标(Evaluation metrics)
对实验 kcat 与预测值计算(原文 Eq. 9–11):
- R²(决定系数):(R^2 = 1 - \frac{\sum_i (y_i - \hat{y}_i)^2}{\sum_i (y_i - \bar{y})^2}),其中 (y_i) 为实验值,(\hat{y}_i) 为预测值,(\bar{y}) 为实验值均值。
- RMSE:(\sqrt{\frac{1}{n}\sum_i (y_i - \hat{y}_i)^2})。
- PCC:预测与实验值的皮尔逊线性相关系数。
段末注释:R² 越大表示解释方差比例越高;RMSE 与 kcat 同量纲,越小越好;PCC 衡量线性相关,不依赖线性校准时仍常用。
3.8 与基线模型的对比协议(Comparison of different deep learning models)
- 选用可公开获取代码与权重的 DLKcat、TurNuP、DLTKcat 等,在相同或论文声明可比的评估流程下复现/运行。
- 序列相似度分层:用 MMseqs2 计算测试集酶与训练集酶之间的序列相似度,将测试样本分为 0–50%、50–90%、90–100% 三档,分别报告 R² 等(原文 Fig. 3c)。
- 结构相似度示例:对案例蛋白用 Foldseek、US-align 等计算结构对齐分数(如 TM-score),与序列一致性对照。
3.9 饱和突变数据上的预测流程(Prediction performance … saturation mutagenesis)
- CYP2C9、PafA 等:从已发表高通量实验文献获取突变体序列与活性/动力学标签;对突变体同样用 ColabFold 预测结构,再输入 DeepEnzyme 得到预测 kcat,与实验分层(错义/无义,或高/低 kcat)比较。
3.10 残基重要性解释(Interpretation analysis … key residue sites)
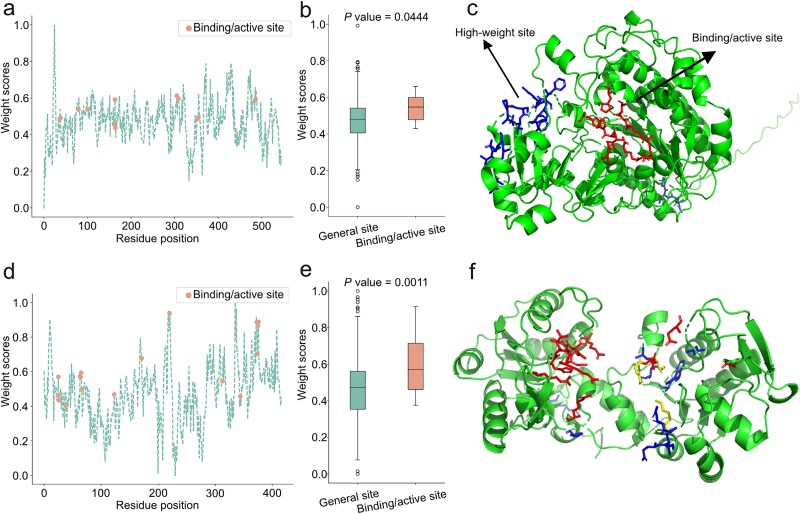
- 对野生型蛋白做 kcat 前向预测时,从 GCN 提取结构支路的残基相关表示,经 min-max 归一化 得到每个残基的权重分数;与 UniProt 中标注的 binding/active site 对比,并考察 Top 5% 高权重残基与注释位点在三维空间中的邻近或重叠程度。
3.11 统计检验与开源实现超参(Statistical analysis & implementation notes)
- 假设检验:组间比较使用 SciPy 中的双侧 t 检验(two-sided t-test)(原文 Methods / Statistical analysis)。
- 仓库示例
example.py中的典型超参(仅供对照,完整训练以作者脚本为准):ngram = 4;dim = 64,hidden_dim1 = 64,hidden_dim2 = 128;Transformer 编码器layers_trans = 3,nhead = 4,hid_size = 64;底物指纹展开 radius = 2;底物与蛋白 GCN 模块在示例中共享同一GCN类实例化参数;dropout在示例推理中可为 0。若论文正文与代码后续更新不一致,以正式发表论文及 Figshare 附带配置为准。
文献原图(Fig. 1):DeepEnzyme 总体框架——Transformer 提取序列特征,GCN 基于接触图与底物邻接矩阵提取结构/底物特征,融合后经注意力读出预测 kcat;图中注明训练用结构由 ColabFold 预测、平均 pLDDT 等。(图源:Wang 等,Brief Bioinform 2024,PMC 11880767,CC BY 4.0)
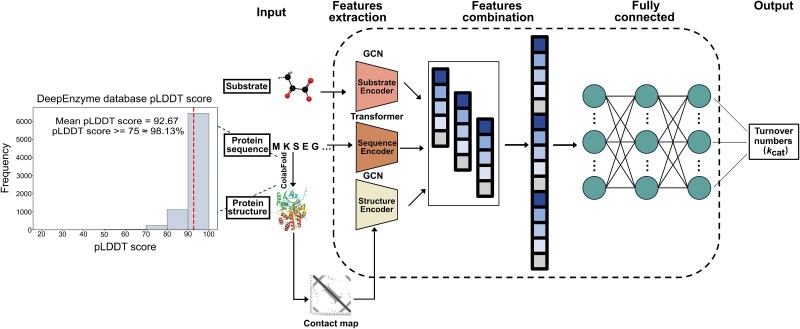
4. 数据与预处理(一览)
已展开见 §3.1:DLKcat 来源、MMseqs2 去冗余(相同底物、序列相似度 > 90% 取最长)、11 927 条组合、80/10/10 划分。补充:Figshare 提供训练用大文件与词典(如 sequence_dict、fingerprint_dict 等),与仓库 Data/Input 路径约定一致。
5. 主要实验结果(概括)
5.1 整体与消融
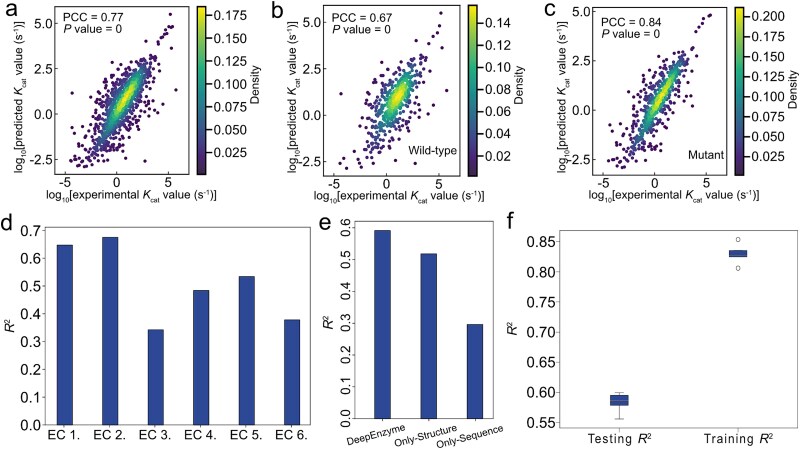
- 测试集上 PCC 接近 0.77(原文 Fig. 2a);五轮训练中测试集平均 R² 约 0.58(Fig. 2f),用于说明不同划分下的稳健性。
- 仅序列 + 底物 与 加入结构 相比,加入结构后精度明显提升(Fig. 2e)。
- 突变体 子集 PCC 高于 野生型(文中约 0.84 vs 0.67),可能与数据集中突变体占比(约 59%)及标签分布有关。
- 按 EC(Enzyme Commission,EC)编号 首位分类时,EC1、EC2 等数据量较大的类 R² 更高,反映类别样本量对性能的影响。
文献原图(Fig. 2):测试集上 PCC、野生型/突变体分层、按 EC 分组的性能、不同输入模态消融(Only-structure / Only-sequence / 全模态)、五轮训练平均 R² 等。(图源同上)
5.2 与既有方法对比
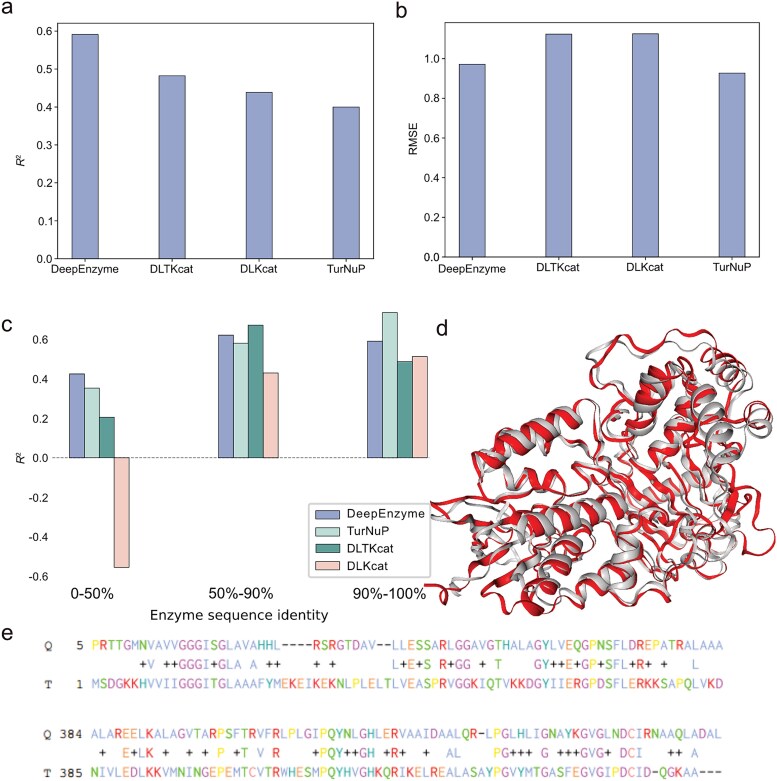
- 与 TurNuP、DLKcat、DLTKcat(可获取代码的公开模型)在相同评估设定下比较:DeepEnzyme 在使用序列 + 结构 + 底物时 R² 更高;RMSE 约为 0.95,低于 DLKcat、DLTKcat,略高于 TurNuP——作者讨论认为 TurNuP 训练时可能剔除了极端高/低 kcat 反应,从而压低 RMSE,对比时需注意数据过滤策略差异。
- 将测试集按与训练集的序列相似度分为 0–50%、50–90%、90–100% 三档:DeepEnzyme 在低相似度档仍保持较高 R²,而 DLKcat、DLTKcat 的 R² 随相似度下降波动更大;作者用 Foldseek、US-align 展示两例 EC 1.3.3.4 酶:序列相似度约 27%,结构相似度(TM-score 等)约 0.88,说明结构信息可补偿远缘序列。
文献原图(Fig. 3):与 TurNuP、DLKcat、DLTKcat 的 R² / RMSE 对比;不同序列相似度分档上的 R²;低序列相似但结构高相似的两例酶示意图。(图源同上)
5.3 饱和突变与可解释性
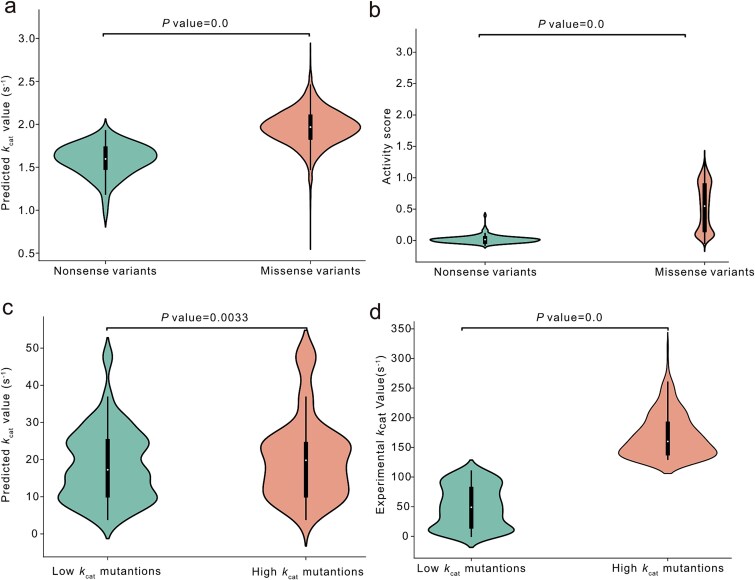
- CYP2C9 大量变异体:无义突变预测 kcat 中位数低于错义突变,与实验活性趋势一致(原文 Fig. 4a–b)。
- PafA 饱和突变:高 kcat 与 低 kcat 突变组的预测中位数差异与实验趋势方向一致(Fig. 4c–d)。
- 对 PafA 与 P00558 等:用 GCN 结构向量经 min-max 归一化 得到残基权重;结合/活性位点(UniProt 注释)权重显著高于一般位点,权重 Top 5% 残基 与注释位点在空间上邻近或部分重叠(Fig. 5),支持模型对功能位点的敏感性。
文献原图(Fig. 4):CYP2C9 变异体预测 kcat 与实验活性分;PafA 突变体高/低 kcat 组的预测与实验对比。(图源同上)
文献原图(Fig. 5):UniProt 注释的结合/活性位点与一般位点的权重分数比较;高权重残基(Top 5%)与功能位点在三维结构中的空间关系(PafA、P00558)。(图源同上)
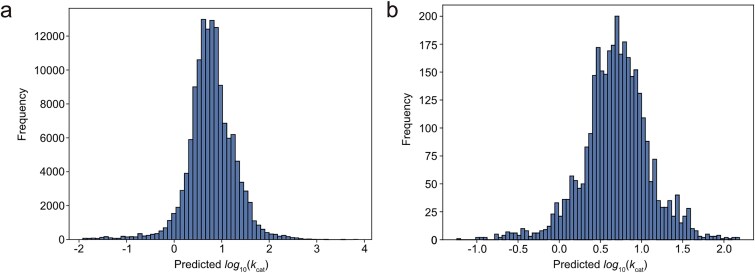
5.4 基因组尺度应用示例
- 对 E. coli、小鼠、酿酒酵母、人 等物种的 GEM 及 Geobacter metallireducens 等模型中的酶促反应做批量 kcat 预测,展示预测值分布(原文 Fig. 6),用于说明组学尺度动力学参数填充的用法。
文献原图(Fig. 6):多物种 GEM 与 Geobacter metallireducens(iAF987)中酶促反应 kcat 预测值的分布。(图源同上)
6. 讨论中的自我定位:与 UniKP 等的关系
作者在 Discussion 中说明:同期 UniKP(预训练 SMILES Transformer + 蛋白质语言模型等)在部分测试上精度可优于 DeepEnzyme;但在单点突变对催化效率的定性排序等任务上 DeepEnzyme 仍有可取之处(见原文 Supplementary Table 1)。同时强调:在结构已预先算好的前提下,DeepEnzyme 参数量更小、对大规模变异体预测速度显著更快(文中举例相对 UniKP 完成相同预测任务的速度数量级差异,依赖具体硬件与实现)。
7. 局限与可改进方向(原文归纳)
- 训练数据规模仍有限(约 1.2 万对),扩大酶–底物多样性有望继续提升性能。
- 未显式纳入 pH、温度 等实验条件,可能与部分酶种预测偏差有关。
- 未来可接入预训练蛋白质/分子表征,在现有 Transformer + GCN 框架上进一步融合。
8. 代码与数据
- 代码:https://github.com/hongzhonglu/DeepEnzyme
- 训练用大文件(Figshare):https://figshare.com/articles/dataset/DeepEnzyme/25771062
- PDF(PMC):https://pmc.ncbi.nlm.nih.gov/articles/PMC11880767/pdf/bbae409.pdf
9. 与酶改造实践的关联(一句话)
若工作流中已具备或愿意批量生成酶三维结构(实验或 ColabFold/AlphaFold 等),DeepEnzyme 代表一条结构增强的 kcat 监督学习路径;与纯序列或预训练大模型路线(如 UniKP)的取舍需综合精度、推理成本、是否需要位点级解释与突变排序来选。