官方资料
ECOLE: Learning to call copy number variants on whole exome sequencing data
ECOLE github
ECOLE 是一款基于 深度学习 进行 全外显子测序(WES)数据中精确检测拷贝数变异(CNVs) 的方法。该方法是基于 Transformer 架构的变体,该模型通过对匹配的 WGS 样本进行高置信度调用,学习调用每个外显子的 CNV。我们通过迁移学习通过一小组专家调用进一步训练和微调模型。
我们证明 ECOLE 首次在人类专家标记数据上实现了高性能,准确率达到 68.7%,召回率达到 49.6%。与排名第二的最佳方法相比,准确率和召回率分别提高了 18.7% 和 30.8%。
软件开发相关的数据
基础训练数据
- 707 samples(WGS & WES) from the 1000 Genomes Project Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426-3440.e19 (2022).00991-6?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867422009916%3Fshowall%3Dtrue)。数据基于Illumina(WES)和NovaSeq(WGS)测序,使用CNVnator 对WGS进行CNV检测,结果作为最终的分析标签,一共标记了740178个DEL 和 953202 个 DUP, 没有标记的的区域作为阴性区域。
不同测序仪的测试数据
- 对 NA12878 样本进行的调用来测试 ECOLE 对各种测序平台的通用性。该样本具有以下平台提供的WES数据:BGISEQ 500、Illumina HiSeq 4000、MGISEQ 2000和NovaSeq 6000。我们仅使用该样本进行测试,其数据不以任何方式包含在训练集中。我们再次使用 CNVnator 调用 NA12878 的 WGS 样本来获取每个外显子的半真实标签以进行评估。此样本总共标记有 1,780 个 DEL 和 1,350 个 DUP 调用。
微调数据
- 15 个 CNV WGS 调用者对来自 1000 个基因组项目的 9 个样本的经过人类专家验证的共识调用。我们获得了8个样本的调用,在1000 Genomes数据集中也有匹配的WES数据,即:HG00512、HG00513、HG00731、HG00732、HG00733、NA19238、NA19239、NA19240。 Chaisson, M. et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 10, 1–16 (2019).
- GiaB 为 Ashkenazi 家族(NA12878 - 儿子、NA12891 - 父亲和 NA12892 - 母亲)提供基于 MetaSV 的 CNV 呼叫集。我们使用 NA12891 的调用对 ECOLE 模型进行了 8 个时期的微调。训练样本总共标记有 10,824 个 DEL 和 1,362 个 DUP 调用。真正的阴性是没有进行调用的其余外显子。我们在为 NA12892 提供的基于 MetaSV 的 CNV 调用上测试了该模型。
- 16 名膀胱癌患者的匹配 WES 和 WGS 样本(登录号:SRP017787)。我们使用该数据集中的 3 个癌症样本(样本 B112、B24、B80)对 ECOLE 模型进行微调 31 。我们使用在这 3 名患者的匹配 WGS 样本上获得的半真实标签进行微调。训练样本总共标记有 23,383 个 DEL 和 282,573 个 DUP 调用。真正的阴性是没有进行调用的其余外显子。Guo, G. et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat. Genet. 45, 1459–1463 (2013).
软件原理介绍
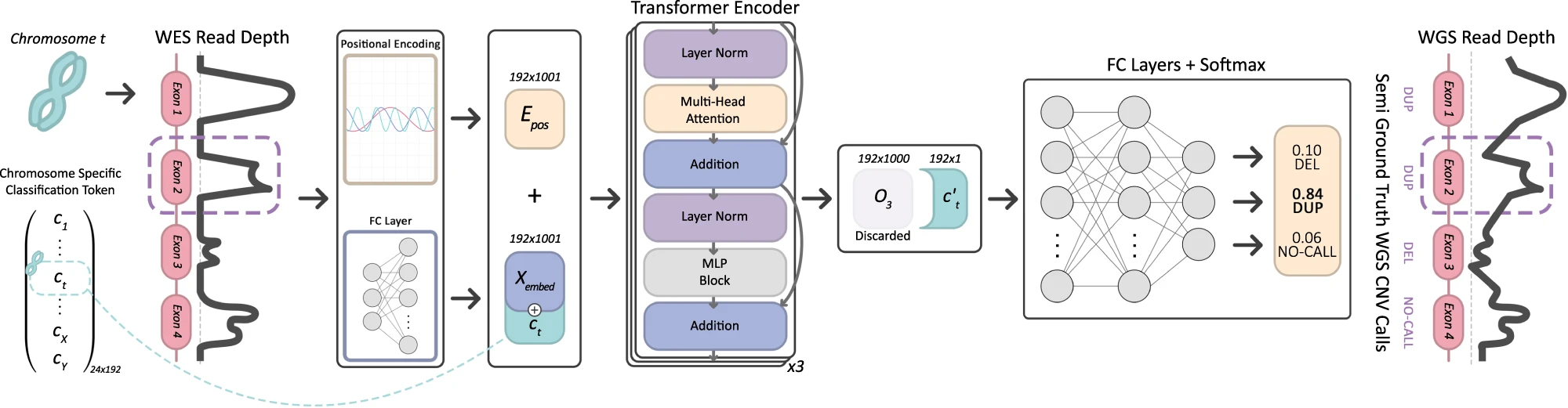
模型输入为每个外显子的:
- readsthe read depth signal (length 1000, padded and masked),
- chromosome number, and
- start and end coordinates of the region.
使用全连接 (FC) 层将每个 1000 个读取深度值映射到更高维度的向量
$${X}{embed}\in \mathbb{R}^{192}$$ (输入嵌入),该层与染色体特定的分类标记向量 $${c}{t}\in \mathbb{R}^{192}$$
对于每条染色体 $t$。这些染色体特定的标记使模型能够学习外显子样本的染色体上下文以执行调用。 Transformer 层使用多头注意力机制,学习碱基对的每个读取深度值相对于给定外显子样本中所有其他碱基对的连接。因此,注意力机制还学习分类标记需要针对相应的 CNV 调用关注哪些读取深度值。为了进一步了解染色体内碱基对的位置上下文,样本的起始和结束坐标用于计算外显子特异性位置编码
$${E}{pos}\in \mathbb{R}^{192\times 1001}$$
两个矩阵被连接并输入到级联的 3 个转换器编码器,生成输出向量
$${O}{3}\in \mathbb{R}^{192\times 1001}$$
然后,获取染色体特定分类标记 $${c}_{t}^\prime$$
的映射变换,其大小为 $$\mathbb{R}^{192}$$
最后,对于最终决策(DEL、DUP 或 NO-CALL),我们使用 2 个 FC 层,然后进行 softmax 激活。
使用说明
安装部署
1 | install conda |
执行使用
1 |
|
参考
#1.