在阅读 AlphaFold(DeepMind 团队的蛋白质结构预测模型体系)或 ESMFold(Meta 团队基于蛋白质语言模型(Protein Language Model,PLM)的折叠模型)给出的 .pdb 结果时,许多疑问集中在「每一列数字是什么」「颜色/粗细是不是置信度」「为什么没有某些残基」。本文从蛋白质结构数据库(Protein Data Bank,PDB)格式入手,说明坐标文件如何编码原子与置信度信息,并穿插两类代表性预测模型的输出习惯,便于把文件内容与生物学解释对上号。
段末注释:PLM 指在大规模序列上预训练、用于表征蛋白质序列上下文的深度模型;PDB 此处主要指 wwPDB 推广的文本坐标格式规范(同名数据库简称亦为 PDB);后文沿用 PLM、PDB。
精确列宽与字段定义以 wwPDB PDB 格式说明 为准。正文配图使用相对目录 ./fileformat-pdb/文件名.png(与 post_asset_folder 资源子目录一致)。
1. PDB 格式是什么:文本化的「原子清单」
PDB 格式历史上是为晶体学与磁共振结构提交而设计的固定宽度文本格式:每一类信息占用特定类型的「记录行」(record),其中描述三维坐标的最核心是 ATOM 与 HETATM 行——前者多为标准氨基酸/核酸骨架上的原子,后者多为配体、离子、结晶添加剂等非标准化学组分。
与之并列、近年越来越常用的是 宏分子晶体学信息文件(macromolecular Crystallographic Information File,mmCIF) 扩展名常为 .cif,更适合超大Assembly、多链 metadata 与自动化校验;**AlphaFold 蛋白质结构数据库(AlphaFold DB)**批量分发时常同时提供 mmCIF 与 PDB。若只做可视化或小脚本解析,PDB 仍极常见;若做数据库对接或宏装配体,应优先熟悉 mmCIF。
段末注释:mmCIF 是基于 STAR 语法的键–值表格化文本格式,一条目可多表关联;后文沿用 mmCIF。
2. 一条 ATOM 行长什么样(列字段一览)
标准 PDB 的 ATOM 行按列位置(column)切片,而不是按空格 split() 可靠解析——因为短字段可能不带前置空格。下图给出「从左到右」的直觉分区(具体列号见后表)。
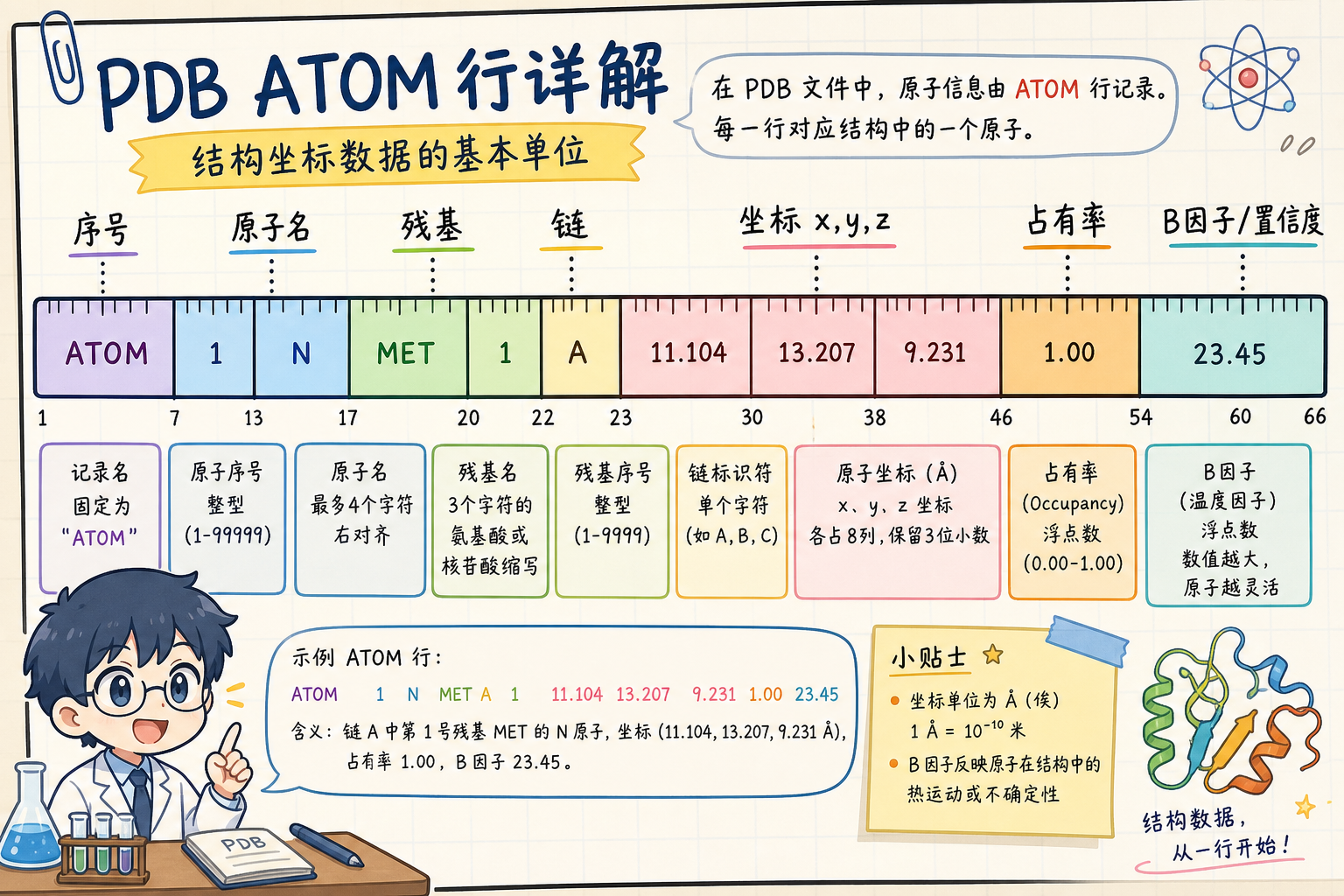
2.1 常用列位(理解预测结构时的重点)
以下列号约定遵循 wwPDB PDB 格式描述中 ATOM 记录的典型布局(不同教程排版可能差 1 列,以官方文档为准):
| 列区间(约) | 字段名(英文) | 含义(阅读预测结构时) |
|---|---|---|
| 7–11 | serial | 原子序号,Unique within model |
| 13–16 | atom | 原子名,如 CA 为 Cα,N / C / O 等 |
| 17 | altLoc | 交替构象代码;预测结构里常为空白 |
| 18–20 | resName | 残基三字母,如 ALA、GLY |
| 22 | chainID | 链标识 A、B… |
| 23–26 | resSeq | 残基序号(作者或预测管线编号) |
| 27 | iCode | 插入码(insertion code),常为空白 |
| 31–38 | x | X 坐标(Å) |
| 39–46 | y | Y 坐标(Å) |
| 47–54 | z | Z 坐标(Å) |
| 55–60 | occupancy | 占有率;晶体学里常见 <1;预测结构里常为 1.00 |
| 61–66 | tempFactor | 温度因子 / B-factor 栏位;在 AlphaFold/ESMFold 输出中常被写入残基级置信度标量 |
| 77–78 | element | 元素符号,如 C、N、O、S |
读预测 PDB 时最值得盯的两列:resSeq + chainID(你在序列比对、突变映射时用);tempFactor(可视化软件里常映射为 B-factor putty 或按残基着色)。
2.2 知识补充:占有率(Occupancy)与温度因子(B-factor)
表格里的 **occupancy(占有率)**与 tempFactor(温度因子,常称 B-factor / B 值)来自晶体学精修传统;读懂其理化意义,有助于区分「实验结构里的无序/热运动」与「预测结构里借用字段存置信度」两种语境。
占有率(Occupancy)
在单晶 X 射线衍射得到的电子密度图中,密度是全晶体、长时间、大量分子的平均。若某一原子位点在两种(或多种)互斥构象之间互换——例如侧链两种翻转、配体两种取向——用一套坐标往往拟合不好密度,精修中就会为同一化学位点建模多套坐标(常与 altLoc 交替构象码配合),并为每套赋予 0~1 的占有率,表示「平均结构里该构象所占比例」,多套占有率之和通常为 1。
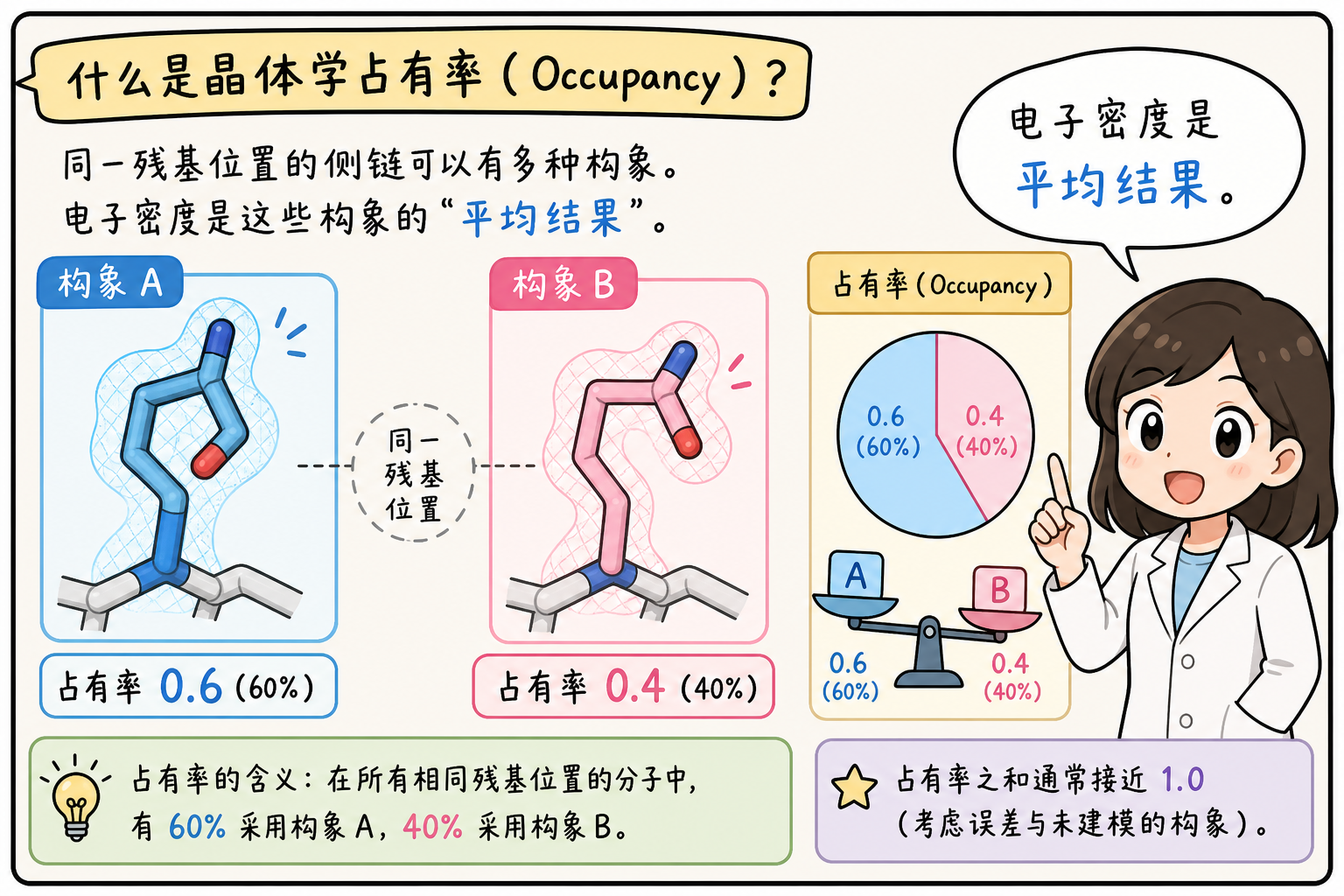
要点:占有率描述的是对平均电子密度的建模权重,与动力学「概率」有类比,但并非轨迹直接测得的概率。预测 PDB 中占有率常为 1.00,多作格式占位,不表示已完成晶体学意义上的无序分解。
段末注释:**交替构象(alternate conformation)**指同一残基/位点在晶体中呈现可区分的多套坐标,用
altLoc字母区分。
温度因子(B-factor / Debye–Waller)
B-factor 刻画原子在平均平衡位置附近的弥散程度:数值越大,电子密度在实空间中显得越「糊」、原子有效越「胖」。来源包括热振动(真随温度升高的位移涨落)与静态无序(不同晶胞中该原子平均位置略有差异,叠加后仍显弥散),实验结构中二者常不可分。各向同性模型用标量 B_iso;更精细时用 各向异性位移参数(anisotropic displacement parameters,ADP) 描述椭球形涨落。
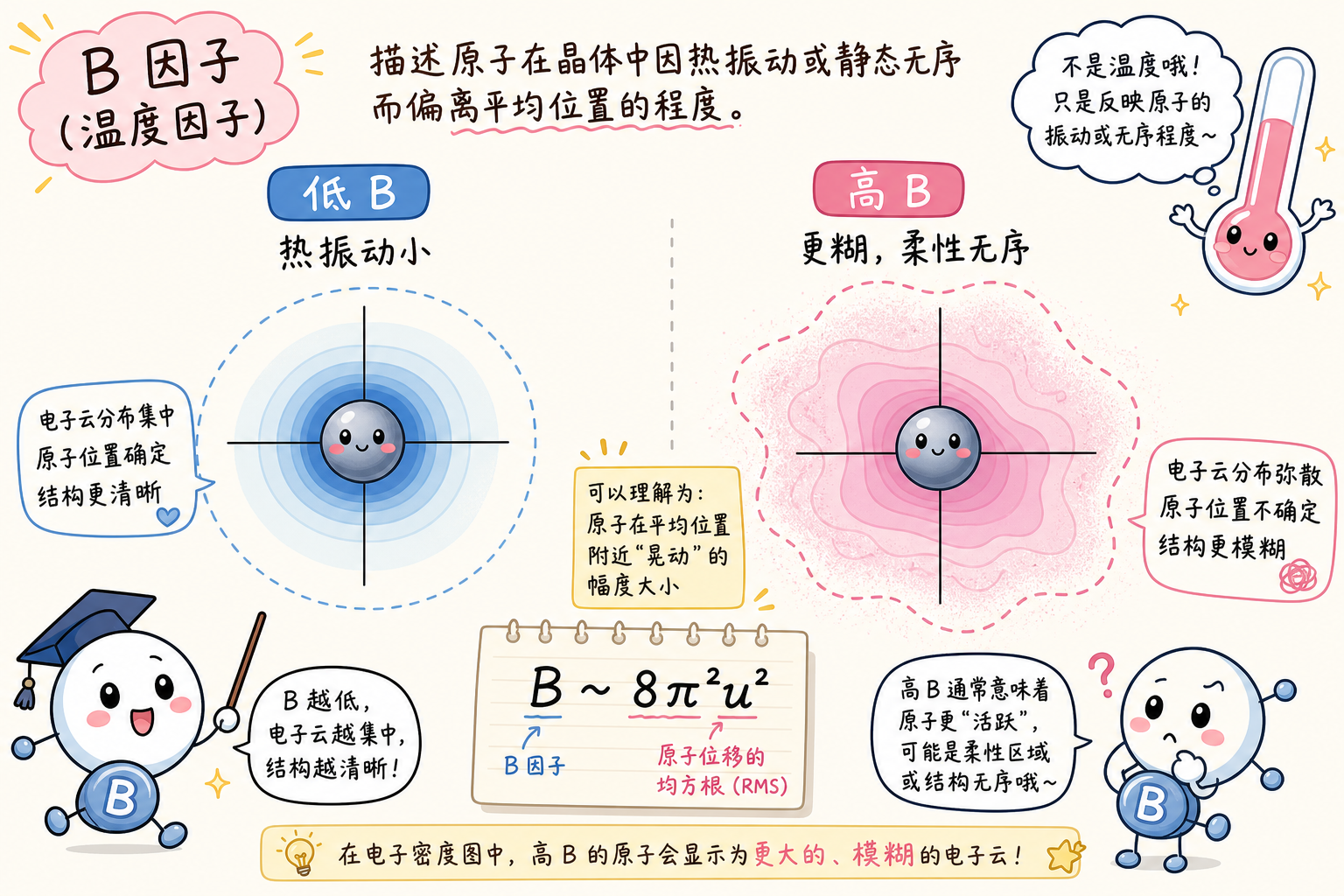
与化学/功能直觉:低 B 多见于刚性内核、主链氢键网中的原子;高 B 常见于表面环、柔性尾巴、弱有序配体或溶剂附近,常与构象可塑性相关,但不能单靠 B 值下结论,需结合密度图、重复结构或分子动力学等。
预测结构中的 tempFactor 列:如 AlphaFold / ESMFold 常把 pLDDT 等置信度写入 B-factor 栏位,属于借用字段名;此时物理含义已不是晶体学精修中的热位移/无序分解结果,可视化时应按软件说明当作置信度映射,勿与实验 B 值直接数值对比。
段末注释:Debye–Waller 因子与 B 在晶体学语境中常一起讨论;简化关系可记为 (B \approx 8\pi^2\langle u^2\rangle)((\langle u^2\rangle) 为均方位移,单位与常数约定需与文献一致)。
3. 其它常见记录行(扫文件时会在头部/尾部看到)
| 记录类型 | 作用(直觉) |
|---|---|
HEADER / TITLE |
条目标题类信息;预测结构里可能有服务器或模型名 |
REMARK |
注释;AlphaFold 常用 REMARK 说明 pLDDT 存放位置、版本等 |
SEQRES |
按链给出的序列(若有);预测结构不一定完整等价于输入 FASTA |
MODEL / ENDMDL |
多模型坐标包裹(NMR 传统);部分管线亦可用于多模型ensemble |
TER |
链终止 |
END |
文件结束 |
若某些残基未建模,实验结构里可能在 REMARK 465 等备注中声明缺失;预测结构也可能在备注或单独 JSON/日志里说明被裁剪区段,需要一并阅读。
4. 结合 AlphaFold:从序列到 PDB,置信度怎么进文件?
AlphaFold 2(AlphaFold 第二代公开架构)在竞赛与后续服务中输出的坐标,普遍将预测局部距离差分检验(predicted Local Distance Difference Test,pLDDT)以每个原子的 tempFactor 字段重复同一残基分值的方式写入 PDB(同一残基内各原子通常共享该残基的 pLDDT)。可视化工具(如 PyMOL、ChimeraX、Mol*)常默认把 B-factor / temperature factor 映射成**从蓝(高置信)到红(低置信)**的配色。
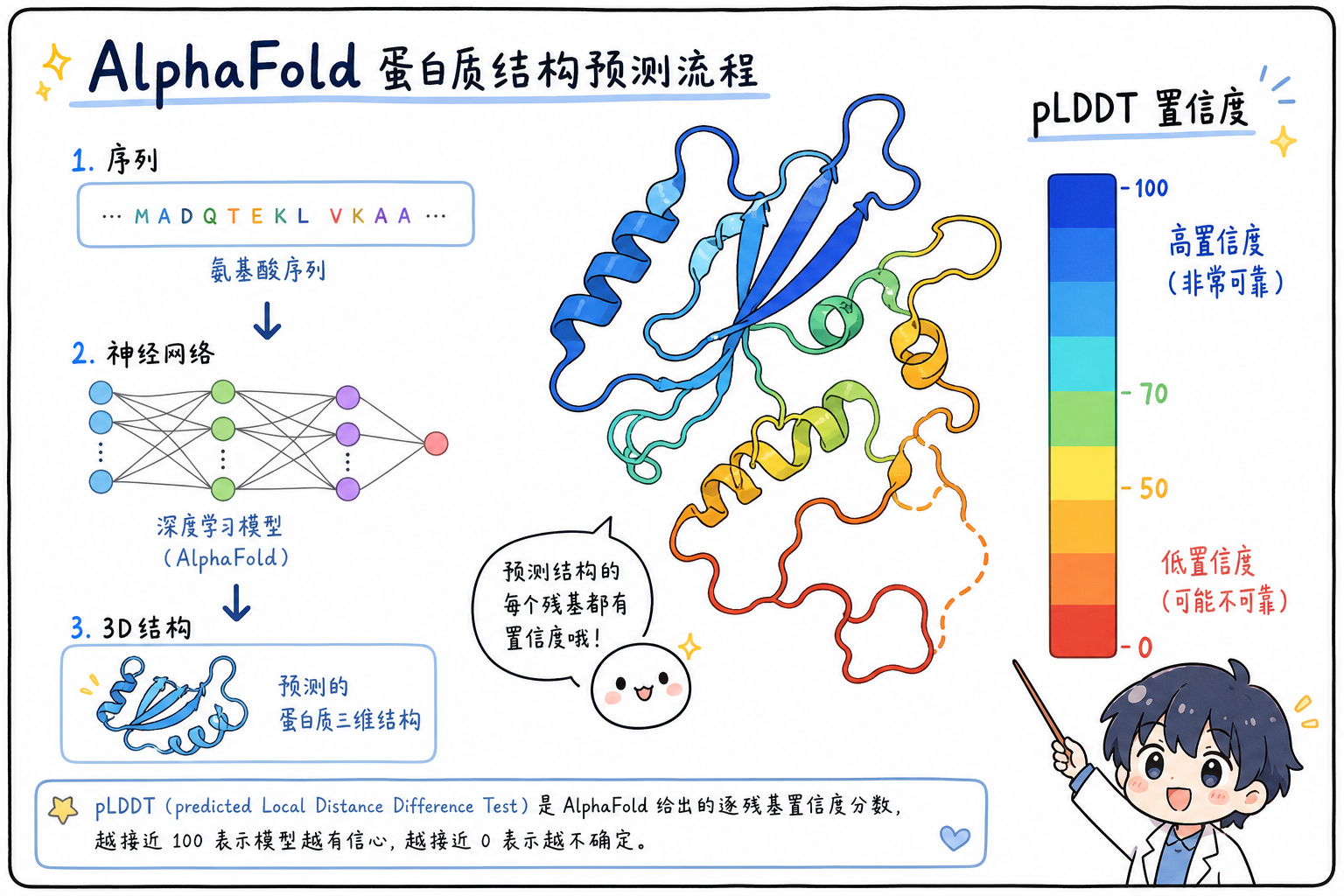
阅读建议:
- 全链着色:快速找柔性环区、无序尾、结构域接口等低分片段。
- 与数据库条目对照:AlphaFold DB 条目除坐标外,网页端还提供 PAE(predicted Aligned Error,预测对齐误差矩阵)等,PAE 不在标准 PDB 单行里,需从配套 JSON 或 mmCIF 扩展字段获取。
- AF3 / 服务器新版本:输出形态可能以 mmCIF 为主或附带更多配体/核酸字段;遇到
.cif时应用支持 mmCIF 的库(如 Biopython 的MMCIF2Dict、gemmi 等)解析。
段末注释:pLDDT 为 0–100 的标量置信度指标,越高表示该残基周围局部几何越可信;PAE 描述残基对之间的预测误差预期,用于评估结构域相对取向是否可靠。
5. 结合 ESMFold:单序列推理与 PDB 字段习惯
ESMFold 使用在亿级序列上训练的 ESM-2 等 PLM 表征,可在仅提供单条序列时较快给出全链坐标。其公开发布的 PDB 类结果,同样常见 tempFactor 承载置信度的惯例(具体标度请以对应用户文档与版本为准),便于与 AlphaFold 流程在可视化层面接轨。
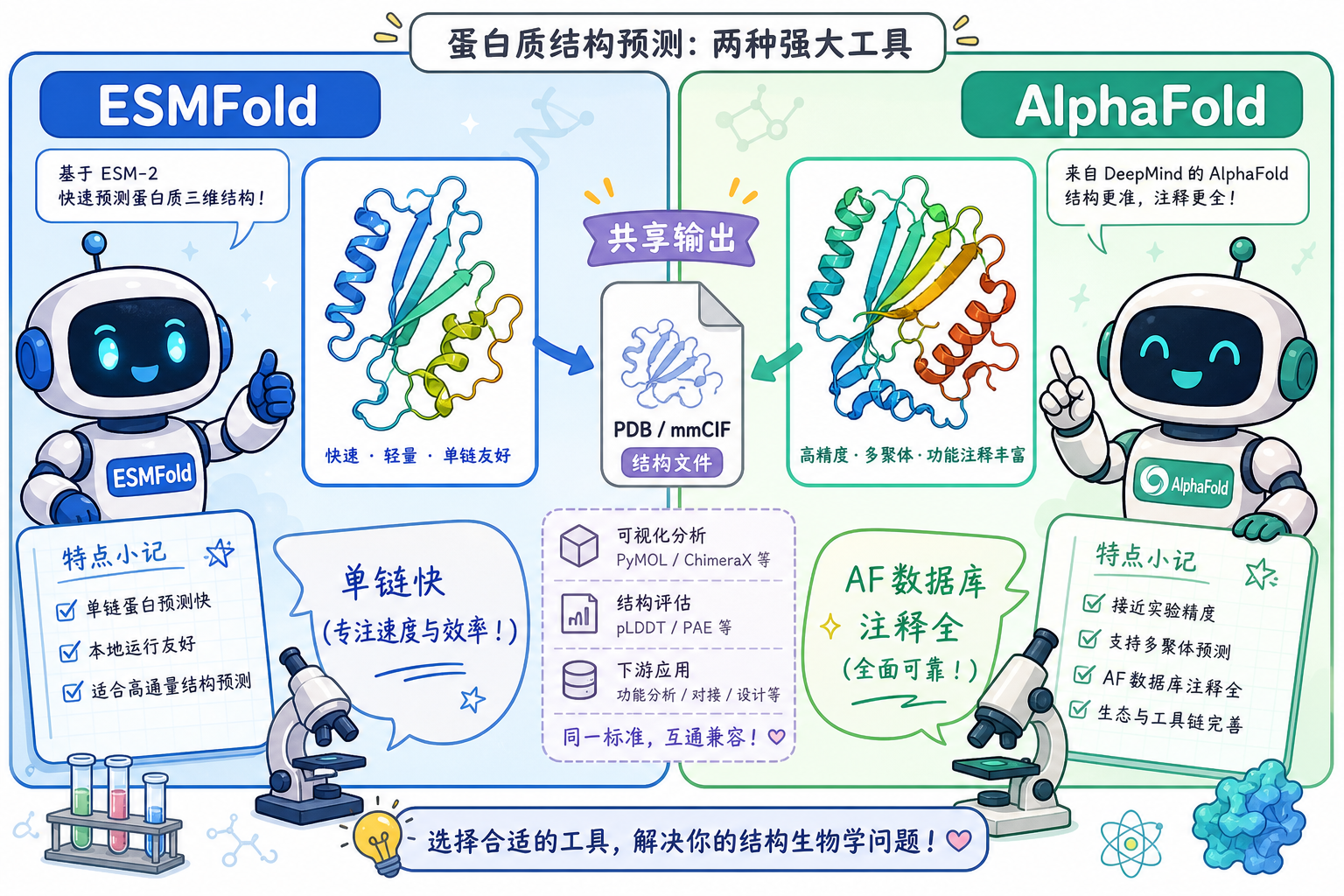
实践对比(概念层,非性能排名):
| 维度 | AlphaFold 体系(典型认知) | ESMFold(典型认知) |
|---|---|---|
| 输入 | 常配合 MSA(multiple sequence alignment,多序列比对)信息更充分 | 强调单序列即可推理,延迟较低场景多 |
| 输出文件 | PDB / mmCIF + 丰富元数据(视版本与入口) | PDB 等;置信度字段解读需查对应版本说明 |
| 解读重心 | pLDDT + PAE + 数据库注释 | 同样关注 B-factor 着色与低分区段生物学含义 |
段末注释:MSA 指将同源序列对齐成矩阵,以共进化信息辅助接触预测与结构约束。
6. 最小示例:伪代码式的一小段 ATOM
下面为教学用虚构片段(列对齐仅为示意,实际文件须严格列宽):
1 | ATOM 1 N MET A 1 12.345 23.456 34.567 1.00 85.12 N |
此处若来自 AlphaFold 类输出,85.12 一列可能对应 pLDDT≈85 的残基置信度(具体是否完全相同取决于导出脚本是否做缩放)。
7. 解析与可视化:为何不推荐 naive split
- 不要用简单按空格切分行再按下标取列——遇到右对齐字段时会错位。
- 推荐:使用成熟解析库(Biopython
PDBParser、gemmi、Open Babel 等),或严格按 wwPDB 列号切片。 - 可视化:在 PyMOL 等软件中学习
cartoon、color b、B-factor putty 等命令,把tempFactor映射到颜色或管径。
8. 小结 Checklist(拿到预测 PDB 时)
- 确认文件是 PDB 还是 mmCIF,选用对应工具链。
- 用
chainID+resSeq做序列/突变映射。 - 用
tempFactor(B-factor) 看置信度着色,结合REMARK;区分预测写入的置信度与实验晶体学 B 值的语义。 - 阅读实验结构时,注意
occupancy小于 1、altLoc与多套坐标所暗示的无序/多构象;预测结构里占有率常为 1.0,勿过度解读。 - 需要评估结构域间取向时,补充查看 PAE(若提供),勿仅凭单残基 pLDDT。
- 注意 缺失残基、插入码、非标准配体在
HETATM/备注中的声明。
9. 小分子配体与酶改造注释中的交换格式
酶改造中与 分子对接、药效团或 辅因子/底物类似物相关的三维结构,除写在 HETATM 中与酶一并存档外,常另存 Molfile、SDF、MOL2 便于对接批量复现与高通量注释;格式要点见同目录 fileformat-molfile.md、fileformat-sdf.md、fileformat-mol2.md。
参考与延伸阅读
- wwPDB PDB Format Description
- AlphaFold DB
- AlphaFold 方法论文与补充材料(Nature 2021 等版本)
- ESMFold / ESM-2 官方论文与代码仓库说明